Boateng, A. Ramakrishna, I. Hattori, T.

Trimethoxysilane-Mediated Peptide Bond Formation from Unprotected Amino Acids and Amino Acid t-Butyl Esters
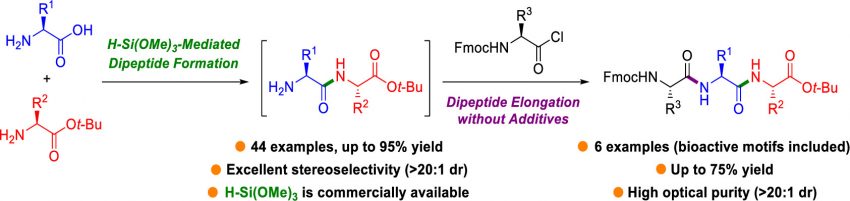
Although the conventional approach to peptide synthesis has been employed for decades, it requires multiple protection–deprotection steps, stoichiometric amounts of coupling reagents and additives, and large volumes of solvent. Consequently, it is accompanied by limitations, such as low atom economy and waste generation. One favorable alternative to combating this problem is the direct formation of peptide bonds using unprotected amino acids. However, this strategy poses issues, such as the low solubility of the unprotected amino acids in organic solvents, unwanted side reactions, and racemization. To overcome these drawbacks, a method has been developed for direct peptide bond formation using trimethoxysilane, an inexpensive and commercially available reagent. Trimethoxysilane helps solubilize the unprotected amino acids in organic solvents, transiently protects the amine group, and simultaneously activates carboxylic acids for coupling. This method represents a one-pot strategy for the synthesis of N-terminal free dipeptides from unprotected amino acids and amino acid tert-butyl esters at 70 °C for 20 h in moderate to good yields (up to 95% yields). Notably, the reactions exhibit high stereoselectivities of >20:1 dr with broad substrate scope compatibility, and the developed approach was applied in the synthesis of tripeptides including bioactive motifs.
Alex Boateng*, Isai Ramakrishna, Tomohiro Hattori*, Hisashi Yamamoto*
J. Org. Chem., 2026, 91, 2530-2537.
Ishihara, K. Hattori, T.

Peptide Bond Formation through Fragment Condensation with Silylating Reagents
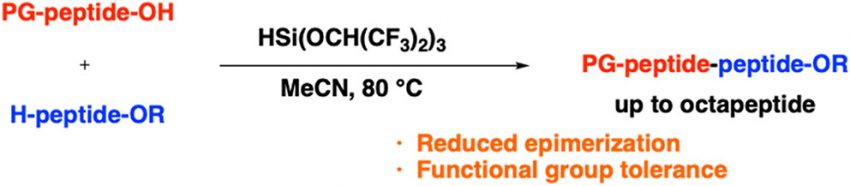
In the context of peptide bond formation, the condensation of peptide fragments facilitates parallel synthesis, thereby streamlining the process and representing an efficient strategy that is unique to liquid-phase chemistry. However, this approach often results in epimerization of the stereocenter adjacent to the activated carboxylic acid due to the formation of oxazolones. The epimerization-free formation of peptide bonds is therefore crucial for the synthesis of peptide drugs. In this study, peptide bonds were formed between peptide fragments with minimal epimerization using HSi(OCH(CF3)2)3 as the silylating reagent. This method was applicable to various peptides bearing diverse functional groups and enabled the synthesis of octapeptides. This strategy holds potential for application in pharmaceutical drug development and in the synthesis of bioactive natural products.
Kotaro Ishihara*, Tomohiro Hattori*, Hisashi Yamamoto*
J. Org. Chem., 2025, 90, 17972-17978.
Hattori, T. Matsunaga, Y.

Additive-Free Peptide Synthesis Using Pentafluorophenyl Esters as the Sequence Oligopeptide Synthesis via a Flow Reaction
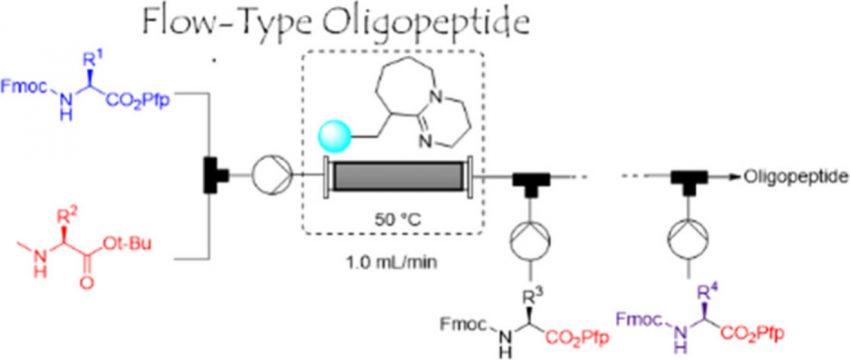
An effective and sustainable protocol for synthesizing oligopeptides via flow chemistry is established herein. Pentafluorophenyl (Pfp) esters readily guide the formation of the desired peptide segments in the presence of nearly equal amounts of nucleophilic amino acid esters as substrates. Although the amino acid Pfp esters are stable, they enable rapid completion of the reactions with nucleophilic amino acids, even with N-methyl amino acid esters. Unlike traditional methods for producing oligopeptides, our process avoids the use of additives and the formation of deficient peptides and other byproducts. To further demonstrate the advantages of the reaction protocol, a flow system was developed using a cartridge filled with 1,8-diazabicyclo[5.4.0]undec-7-ene polymer to facilitate Fmoc-group deprotection. The continuous flow reaction enables sequential peptide elongation to form oligopeptides with high purities and yields. Furthermore, gram-scale synthesis via long-term automated operation is successful, and the continuous-flow system can have ever-increasing practical applications. This powerful methodology should provide solutions to many long-standing problems in organic synthesis, e.g., formation of byproducts and involvement of multiple steps.
Tomohiro Hattori*, Yuki Matsunaga, Hisashi Yamamoto*
Org. Process Res. Dev., 2025, 29, 2815-2822.
Ramakrishna, I. Boateng, A. Hattori, T.

Tris(2,2,2-trifluoroethoxy)silane-Enabled Peptide Bond Formation between Unprotected Amino Acids and Amino Acid t-Butyl Esters
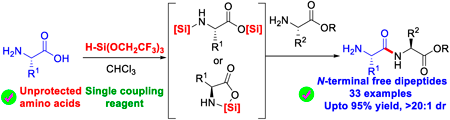
Conventional peptide synthesis involves multiple protection and deprotection steps, and typically relies on stoichiometric amounts of coupling reagents and additives. This makes the process cumbersome, and results in poor atom economy and hazardous waste generation. Therefore, direct peptide bond formation using unprotected amino acids is a promising alternative. However, this approach presents some challenges: 1) Solubility of unprotected amino acids in organic solvents; 2) Control of undesired side reactions; 3) Chemo-selective activation of the carboxylic acid group in the presence of an amine functionality; and 4) Epimerization. To address these challenges, we developed tris(2,2,2-trifluoroethoxy)silane [H-Si(OCH2CF3)3], a cost-effective and accessible coupling reagent. This single reagent efficiently synthesizes N-terminal free peptides from unprotected amino acids and amino acid tert-butyl esters, without the need for any additives. H-Si(OCH2CF3)3 enhances amino acid solubility through coordinating with both termini and plays a dual role, serving as a transient amine-protecting group and as a carboxylic acid activating or promoting reagent for peptide bond formation. This method is operationally simple and versatile, enabling the efficient synthesis of N-terminal free peptides from unprotected amino acids and amino acid tert-butyl esters, with good yields, high optical purity, and broad side-chain compatibility.
Isai Ramakrishna*, Alex Boateng, Tomohiro Hattori*, Hisashi Yamamoto
Chem. Pharm. Bull., 2025, 73, 787-792.
Hattori, T. Kon, K.
Controlled Boc-Protection of Diketopiperazine Using Tributylphosphine
A method for chemoselective acylation of the vacant amide of diketopiperazines (DKPs) using di-tert-butyl dicarbonate (Boc2O) was developed. Nucleophilic tributylphosphine reacts immediately with acid anhydride to form an active quaternary phosphonium cation. This intermediate induces acylation of the amide group on the empty side, resulting in chemoselectivity that could not be controlled by using N,N-dimethyl-4-aminopyridine (DMAP). This method achieved high yields of mono-acylated DKPs, avoiding the acylation of amino acids with bulky substituents. This reaction system also provides chemoselectivity based on the bulkiness of the protecting groups of active functional groups at the side chain, and enhances the range of substrate applicability by achieving acylation of the amido groups of amino acids, except for glycine. Furthermore, using the substrates prepared here, elongation reactions induced by introducing amino acid esters associated with ring opening were also successfully demonstrated under reaction conditions without any additives, highlighting the possibility to form peptides with a broad range of sequences.
Tomohiro Hattori*, Kazumasa Kon*, Hisashi Yamamoto
Chem. Pharm. Bull., 2025, 73, 658-662.
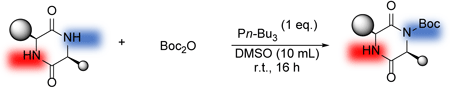
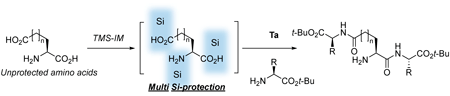
Hattori, T. Watanabe, H. Matsunaga, Y.

Tantalum-Catalyzed Peptide Elongation of Unprotected Amino Acids Using N-Trimethylsilylimidazole
Unprotected amino acids are completely insoluble in organic solvents, limiting their use in organic synthesis. To address this issue, we report a Ta-catalyzed strategy for the elongation of unprotected amino acids using N-trimethylsilylimidazole. More specifically, silylation of the unprotected amino acids was carried out at both termini to generate a linear intermediate that was soluble in organic solvents, thereby permitting the efficient introduction of amino acid esters at the C-terminus. Furthermore, the in situ silicon protection approach employed herein was applicable to varying lengths of amino acid chains, leading to the generation of N-unprotected β-, γ-, and δ-peptides without any loss of enantio- or diastereopurity. These innovative results have facilitated the elimination of several steps in the preparation of desired peptides and have simplified the synthesis of peptides containing long-chain amino acids and branched peptides.
Tomohiro Hattori*, Haruna Watanabe, Yuki Matsunaga, Hisashi Yamamoto*
Chem. Pharm. Bull., 2025, 73, 595-599.
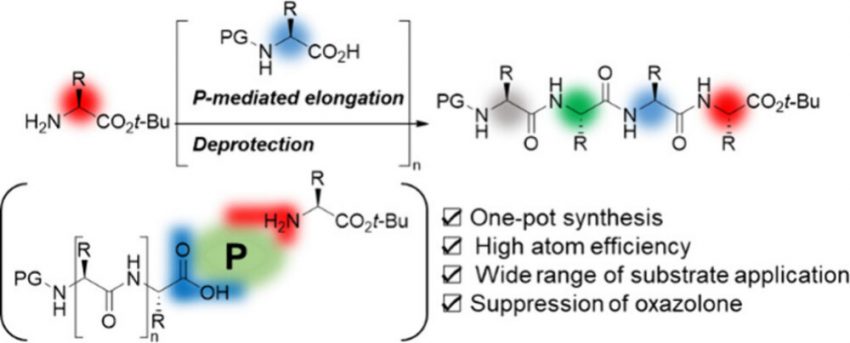
Hattori, T. Ishiguro, T.
Triphenyl Phosphite-Mediated One-Pot Peptide-Bond Formation under Neutral Reaction Conditions
Liquid-phase peptide synthesis requires an excessive amount of strong C-terminal-activating reagents under acidic or basic conditions. Although the reaction proceeds well, it is accompanied by epimerization and other side reactions with active reagents, necessitating stepwise purification. Herein, we present an efficient peptide bond formation using P(OPh)3, which yields the corresponding peptides in high yield, and diastereopurity. In addition, this atom-economical method under neutral conditions minimizes side reactions and facilitates seamless one-pot oligopeptide synthesis.
Tomohiro Hattori*, Tomomi Ishiguro, Hisashi Yamamoto*
Org. Lett., 2025, 27, 3742-3746.
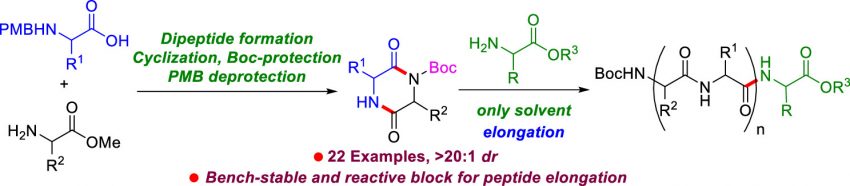
Ramakrishna, I. Boateng, A. Hattori, T. Nakagai, K. Kawase, M. Ogata, S.
Synthesis of Mono-Boc-2,5-Diketopiperazine: A Key Building Block for Amide and Peptide Synthesis
Diketopiperazine (DKP), a versatile scaffold, is extensively used in the synthesis of complex natural products, bioactive molecules, and smart materials in organic chemistry. Recently, activated DKPs, such as Boc-DKPs, have emerged as key building blocks for peptide elongation in peptide synthesis. In this study, we developed a facile protocol for synthesizing mono-Boc-protected DKPs from readily accessible N-4-methoxybenzyl (N-PMB)-amino acids and amino acid methyl esters. This protocol involved a sequence of reactions encompassing the formation of dipeptides from N-PMB-amino acids and amino acid methyl esters, cyclization of N-PMB-dipeptides to form PMB-DKPs, Boc-protection of PMB-DKPs, and subsequent PMB-deprotection of PMB-DKP-Boc to afford mono-Boc-DKPs. The protocol demonstrated a broad substrate scope, accommodating diverse amino acids with various side chains, affording mono-Boc-DKPs in good yields with excellent stereoselectivities (>20:1 dr). The synthetic utility of mono-Boc-DKPs was showcased in peptide synthesis by synthesizing pentapeptide Boc-l-Tyr(t-Bu)-Gly-l-Phe-Gly-l-Val-OtBu by 2-fold peptide elongation with two mono-Boc-DKPs. Furthermore, we synthesized Leu-enkephalin pentapeptide by reacting cyclo(Boc-l-Tyr(t-Bu)-Gly-) with H-Gly-l-Phe-l-Leu-Ot-Bu, resulting in a good yield and excellent optical purity.
Isai Ramakrishna*, Alex Boateng, Tomohiro Hattori*, Kozue Nakagai, Masae Kawase, Shinichi Ogata, Hisashi Yamamoto*
J. Org. Chem., 2025, 90, 4357-4364.
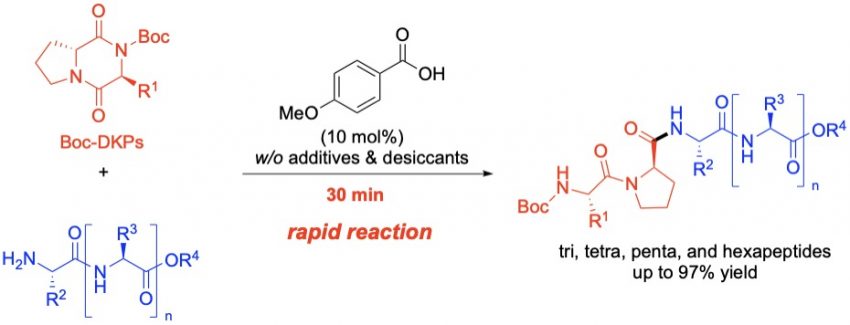
Kon, K.

Carboxylic Acid-Catalysed Transamidation of Boc-Protected Diketopiperazines (Boc-DKPs) for Peptide Synthesis
A carboxylic acid-catalysed transamidation of Boc-protected diketopiperazines (Boc-DKPs) was developed in this study, which allowed access to various tripeptides featuring a D-proline residue in good-to-high yields within 30 min. Subsequent coupling reactions between the Boc-DKPs and di-, tri-, and tetrapeptide esters were successfully performed, furnishing the corresponding tetra-, penta-, and hexapeptides.
Kazumasa Kon*, Hisashi Yamamoto*
Eur. J. Org. Chem., 2025, e202401319
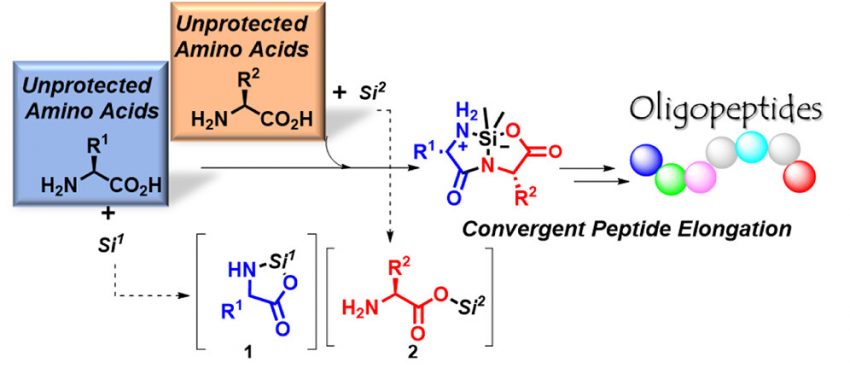
Hattori, T.

Peptide Bond Formation Between Unprotected Amino Acids: Convergent Synthesis of Oligopeptides
In the history of peptide chemical synthesis, amino acid-protecting groups have become basic, essential, and common moieties. As the use of protecting groups for amino acids effectively suppresses unwanted side reactions and enables the desired reaction to proceed selectively, their use continues regardless of the complexities of the protection processes and the waste generated by the deprotection residues. We developed peptide bond formation between unprotected amino acids to form silacyclic dipeptides. This is the first report of the proceeding cross-condensation between an unprotected amino acid and another unprotected amino acid. The selectivity, reaction yields, and purity of the products were satisfactory. In addition, we demonstrated further elongation of these compounds and achieved convergent synthesis with peptide–peptide elongation without the use of coupling reagents. Thus, these methods showed the potential to unlock a new, more efficient synthetic path toward polypeptides.
Tomohiro Hattori*, Hisashi, Yamamoto*
J. Am. Chem. Soc., 2024, 146, 25738-25744
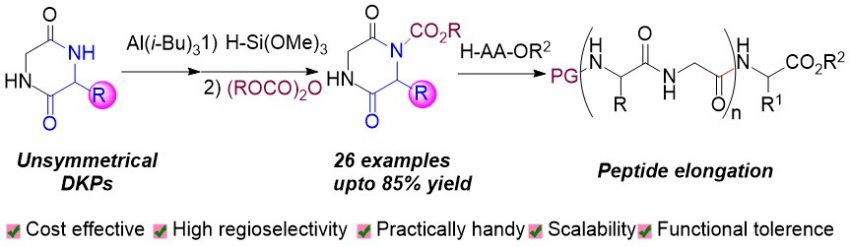
Ramakrshna, I.
Hattori, T.
Ishiguro, T.
Triisobutylaluminium-Mediated Regioselective Protection of Sterically Hindered Amide NH of Cyclo-(AA-Gly): Key Building Block for Next-Generation Peptide Synthesis
In this study, we address the challenge of regioselective Boc protection in the more sterically hindered amide NH of unsymmetrical 2,5-diketopiperazines (DKPs) formed from glycine and various amino acids. Our research introduces a novel technique utilizing cost-effective triisobutylaluminium and trimethoxysilane. Notably, trimethoxysilane selectively reacts with the less hindered amide NH, facilitating the regioselective Boc protection of the more congested amide NH in DKPs. The primary objective of our work is to develop a straightforward and scalable approach for the synthesis of Boc-protected DKPs, with a focus on addressing the steric challenges presented by these compounds. We successfully demonstrate the scalability of this method, enabling the synthesis of a variety of mono-Fmoc, Cbz, Alloc, and EtOCO-protected DKPs. Furthermore, we extend the applicability of this strategy by employing it in the construction of pentapeptides through a twofold-peptide elongation process. Our findings reveal the versatility and efficiency of this regioselective Boc protection method. Overall, this research introduces a valuable solution to the regioselective Boc protection challenge in DKPs and demonstrates its applicability in peptide synthesis, showcasing its potential for further advancements in the field.
Isai Ramakrishna*, Tomohiro Hattori*, Tomomi Ishiguro, Hisashi Yamamoto*
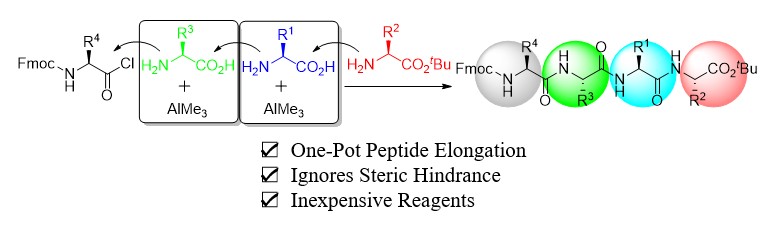
Hattori, T.
Trimethylaluminum-mediated one-pot peptide elongation
Efficient and straightforward peptide bond formation of N-, and C-terminal unprotected amino acids was successfully achieved by using trimethylaluminum. The coupling reaction was accomplished by pre-reacting of N-, and C-terminal unprotected amino acids and trimethylaluminum to form a five-membered ring, that smoothly reacted with nucleophilic amino acid esters. This simple and highly efficient reaction system allows one-pot tripeptide synthesis without the need for the expensive coupling reagents. Furthermore, peptide bond formation can be effectively achieved even for the amino acids with bulky substituent at the side chain to afford the corresponding tripeptides in high yields in a one-pot manner. In addition, the reaction can be applied for further peptide elongation by the subsequent addition of amino acids and trimethylaluminum. We anticipate that this cost-effective, straightforward, and efficient protocol will be useful for the synthesis of a wide variety of peptides.
Tomohiro Hattori*, Hisashi Yamamoto*
Chem. Sci.,2023, 14, 5795-5801.
Wu, A.

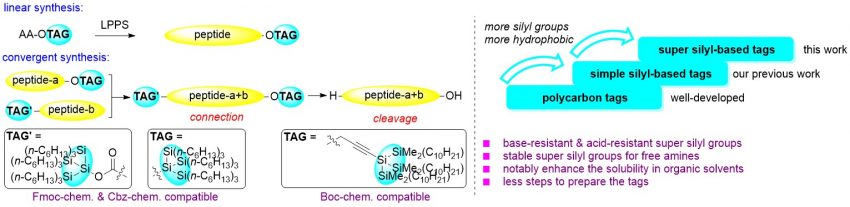
Super silyl-based stable protecting groups for both the C- and N-terminals of peptides: applied as effective hydrophobic tags in liquid-phase peptide synthesis
Tag-assisted liquid-phase peptide synthesis (LPPS) is one of the important processes in peptide synthesis in pharmaceutical discovery. Simple silyl groups have positive effects when incorporated in the tags due to their hydrophobic properties. Super silyl groups contain several simple silyl groups and play an important role in modern aldol reactions. In view of the unique structural architecture and hydrophobic properties of the super silyl groups, herein, two new types of stable super silyl-based groups (tris(trihexylsilyl)silyl group and propargyl super silyl group) were developed as hydrophobic tags to increase the solubility in organic solvents and the reactivity of peptides during LPPS. The tris(trihexylsilyl)silyl group can be installed at the C-terminal of the peptides in ester form and N-terminal in carbamate form for peptide synthesis and it is compatible with hydrogenation conditions (Cbz chemistry) and Fmoc-deprotection conditions (Fmoc chemistry). The propargyl super silyl group is acid-resistant, which is compatible with Boc chemistry. Both tags are complementary to each other. The preparation of these tags requires less steps than previously reported tags. Nelipepimut-S was synthesized successfully with different strategies using these two types of super silyl tags.
An Wu*, Hisashi Yamamoto*
Chem. Sci.,2023, 14, 5051-5061.
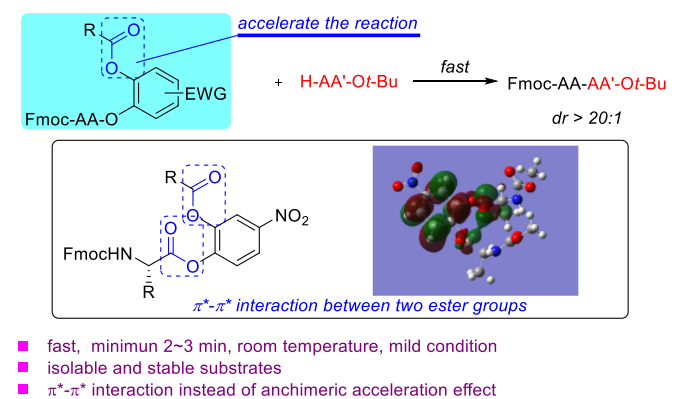
Wu, A.

An Unusual Acceleration of Amination Reactivity by the Proximal Ester Groups in Catechol Diesters: An Efficient Way for Peptide Synthesis
Peptide bond formation has drawn more and more attention due to its importance in medicinal chemistry and drug discovery. Using active esters to construct peptide bonds is an indispensable strategy in this field. We herein report a fast and efficient amination reaction to synthesize peptides from catechol diesters. Catechol monoesters are good active esters in peptide synthesis due to the anchimeric assistance effect. In previously reported work, the amination reaction was inactivated if the 2-hydroxyl group in the catechol esters was functionalized. However, we found an unusual acceleration of the amination reactivity when the 2-hydroxyl group was functionalized as an ester group. The reaction can be complete in 2~3 min in some cases. This acceleration was found through experimental studies to not result from the anchimeric assistance effect. We proposed a π*-π* interaction pathway between the two proximal ester groups. Some computational studies were made to support this assumption.
An Wu*, Hirotaka Ikeda*, Hisashi Yamamoto*
Precision Chemistry, 2023, 1, 100-106
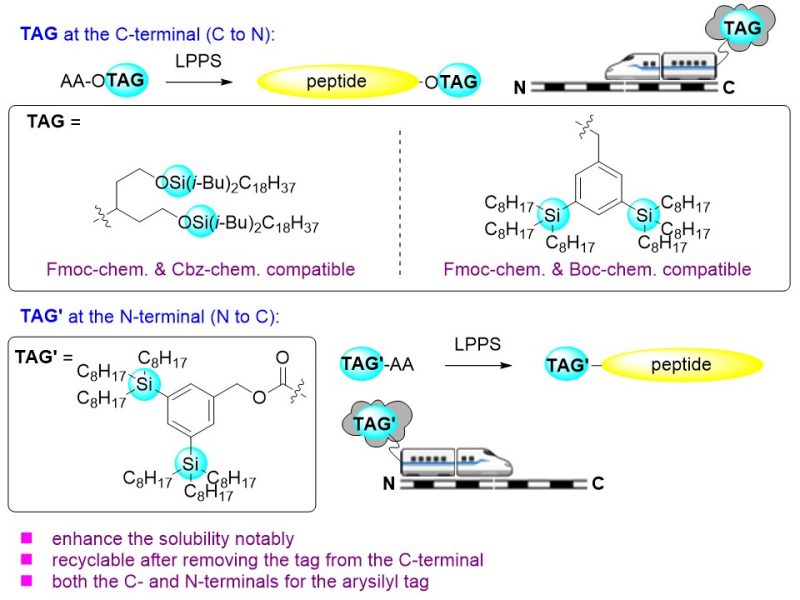
Wu, A. Ramakrishna, I. Hattori, T.
Silicon-based hydrophobic tags applied in liquid-phase peptide synthesis: protected DRGN-1 and poly alanine chain synthesis
Two new recyclable silicon-based hydrophobic tags were developed for installing them at the C-terminal of peptides to increase the solubility in organic solvents and the reactivity of the peptides during liquid-phase peptide synthesis (LPPS). They comprise a siloxy group containing tag and an arylsilyl group containing tag. The hydrophobicity of these tags is much greater than those of previously reported examples. The siloxy group containing tag is compatible with Fmoc-deprotection conditions (Fmoc-chemistry) and hydrogenation conditions (Cbz-chemistry), while the arylsilyl group containing one is resistant to Fmoc-deprotection conditions (Fmoc-chemistry) and Boc-deprotection conditions (Boc-chemistry). Using the siloxy group containing tag, protected DRGN-1, a peptide containing 14 amino acid residues was prepared successfully by using linear synthesis combined with one convergent synthesis step. Using the arylsilyl group containing tag, a poly alanine chain containing 7 alanine residues was synthesized. The arylsilyl group containing tag can also be installed at the N-terminal to elongate the peptides from the N-terminal to the C-terminal. The yields and the solubility of the products in organic solvents in each step were good to excellent with the assistance of these silicon-based tags.
An Wu*, Isai Ramakrishna, Tomohiro Hattori, Hisashi Yamamoto*
Org. Biomol. Chem., 2022, 20, 8685-8692
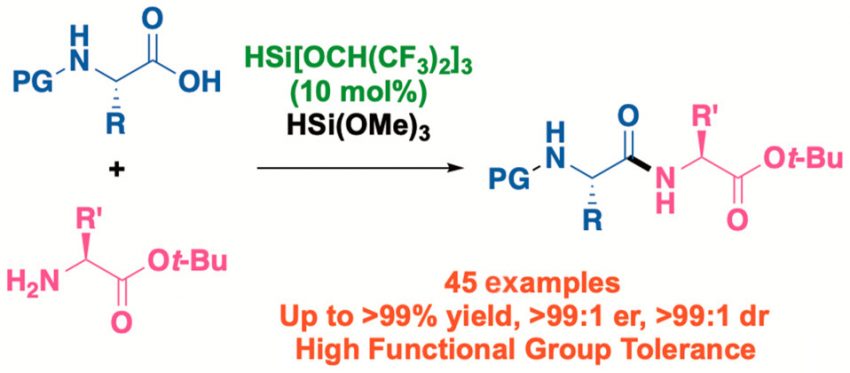
Muramatsu, W.

Organocatalytic Activation of Inert Hydrosilane for Peptide Bond Formation
We describe the development of a reliable catalytic protocol for peptide bond formation that is generally applicable to natural and unnatural α-amino acids, β-amino acids, and peptides bearing various functional groups. A 10 mol % loading of HSi[OCH(CF3)2]3 as a catalyst was sufficient to guarantee a consistently high yield of the resulting peptide. This method facilitates the sustainable utilization of natural resources by using a catalyst and an auxiliary based on earth-abundant silicon.
Wataru Muramatsu*, Hisashi Yamamoto*
Org. Lett., 2022, 24, 7194-7199.
Muramatsu, W.
An economical approach for peptide synthesis via regioselective C–N bond cleavage of lactams
An economical, solvent-free, and metal-free method for peptide synthesis via C–N bond cleavage using lactams has been developed. The method not only eliminates the need for condensation agents and their auxiliaries, which are essential for conventional peptide synthesis, but also exhibits high atom economy. The reaction is versatile because it can tolerate side chains bearing a range of functional groups, affording up to >99% yields of the corresponding peptides without racemisation or polymerisation. Moreover, the developed strategy enables peptide segment coupling, providing access to a hexapeptide that occurs as a repeat sequence in spider silk proteins.
Wataru Muramatsu*, Hisashi Yamamoto*
Chem. Sci., 2022, 13, 6309-6315
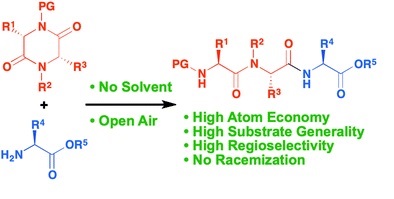
Hattori, T.
Synthesis of Silacyclic Dipeptides: Peptide Elongation at Both N- and C-Termini of Dipeptide
A new type of peptide bond formation utilizing silacyclic amino acids or peptides is described. This work has the following advantages: (1) imidazolylsilane is a highly fascinating coupling reagent for dipeptide synthesis from N-,C-terminal unprotected amino acids with amino acid tert-butyl esters; (2) deprotection of the tert-butyl ester at the C-terminus and cyclization sequentially proceed depending on reaction conditions to afford novel silacyclic dipeptides; (3) the cyclized products show a remarkable capacity as substrates of peptide elongation because the silacyclic compounds can act as both nucleophiles and electrophiles, and this capacity lead to one-pot site-selective tetra- and oligopeptide syntheses. These innovative advantages will help to simplify classical peptide synthesis significantly..
Tomohiro Hattori*, Hisashi Yamamoto*
J. Am. Chem. Soc, 2022,144, 1758-1765
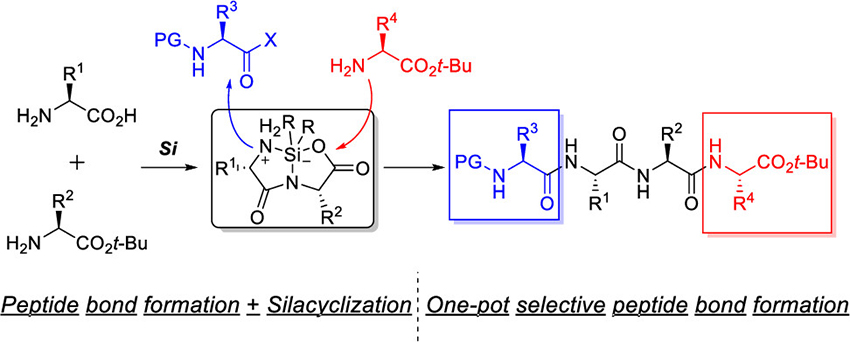
Muramatsu, W. Hattori, T.

Amide bond formation: beyond the dilemma between activation and racemisation
The development of methods for amide bond formation without recourse to typical condensation reagents has become an emerging research area and has been actively explored in the past quarter century. Inspired by the structure of vitamin B12, we have developed a metal-templated macrolactamisation that generates a new wave towards classical macrolactam synthesis. Further, distinct from the extensively used methods with condensation reagents or catalysts based on catalyst/reagent control our metal-catalysed methods based on substrate control can effectively address long-standing challenges such as racemisation in the field of peptide chemistry. In addition, the substrate-controlled strategy demonstrates the feasibility of “remote” peptide bond-forming reaction catalysed by a metal–ligand complex. Moreover, an originally designed hydrosilane/aminosilane system can avoid not only racemisation but also unnecessary waste production. This feature article documents our discovery and application of our original approaches in amide bond formation.
Wataru Muramatsu*, Tomohiro Hattori, Hisashi Yamamoto*
Chem. Commun, 2021, 57, 6346-6359
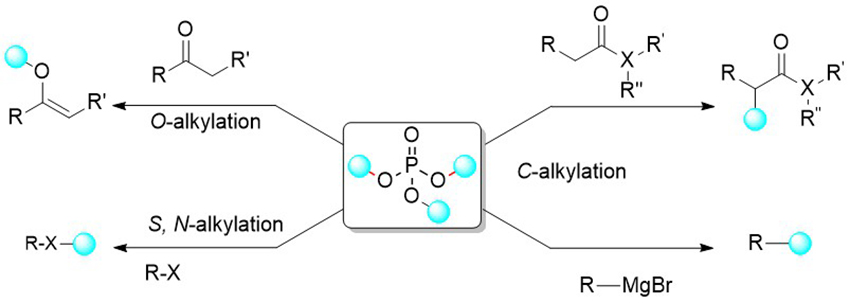
Banerjee, A. Hattori, T.

Regio- and Stereoselective (SN2) N-, O-, C- and S-Alkylation using Trialkyl Phosphates
Bimolecular nucleophilic substitution (SN2) is one of the most known fundamental reactions in organic chemistry to generate new molecules from two molecules. In principle, a nucleophile attacks from the back side of an alkylating agent having a suitable leaving group, most commonly using a halide. However, alkyl halides are expensive, very harmful, toxic and not so stable which makes them problematic for laboratory use. In contrast, trialkyl phosphates are cheap, readily accessible, stable at room temperature, under air, and are easy to handle but rarely used as alkylating agents in organic synthesis. Here, we describe a mild, straightforward and powerful method for nucleophilic alkylation of various nucleophiles such as N-, O-, C- and Susing readily available trialkyl phosphate. The reaction proceeds smoothly with excellent yield and quantitative yield in many cases and covers a wide range of substrates. Further, the rare stereoselective transfer of secondary alkyl groups has been achieved with inversion of configuration of chiral centers (up to >99% ee).
Amit Banerjee*, Tomohiro Hattori*, Hisashi Yamamoto*
SYNTHESIS 2023, 55, 315-332
Muramatsu, W.

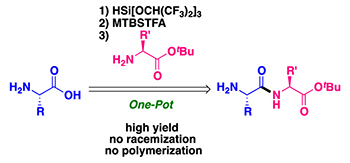
Peptide Bond Formation of Amino Acids by Transient Masking with Silylating Reagents
A one-pot peptide bond-forming reaction has been developed using unprotected amino acids and peptides. Two different silylating reagents, HSi[OCH(CF3)2]3 and MTBSTFA, are instrumental for the successful implementation of this approach, being used for the activation and transient masking of unprotected amino acids and peptides at C-termini and N-termini, respectively. Furthermore, CsF and imidazole are used as catalysts, activating HSi[OCH(CF3)2]3 and also accelerating chemoselective silylation. This method is versatile as it tolerates side chains that bear a range of functional groups, while providing up to >99% yields of corresponding peptides without any racemization or polymerization.
Wataru Muramatsu*, Hisashi Yamamoto*
J. Am. Chem. Soc. 2021, 143, 6792-6797.
Hattori. T, Muramatsu, W

基質支配的アミド結合形成反応:ペプチド合成の新機軸
Peptides, which are elongated of peptides chain of amino acids linked by amide bonds, are elementary components in living systems and regulate many biological processes. Since the solid-phase peptide synthesis was introduced by Merrifield in 1964, chemical synthesis of peptides using solid-phase system have emerged as a valuable method due to its ease of operation and rapid synthesis of desired peptides. However, despite of the effectiveness of the solid-phase approach, typical reaction requires excess amounts of coupling reagents, bases, and amino acids to achieve maximum conversion. And the ease of operation led to accumulate undesired segments together with target peptide because solid-phase system is generally carried out by coupling amino acids with N-terminal amino acid residue of peptides in a stepwise approach without any isolation and characterization at each step. To solve these issues, we focused on the development of catalytic liquid-phase synthesis, which has advantage of isolating the growing peptide chain from reaction solution after each coupling step. Although several liquid-phase methods have been developed, there is a still considerable room for the improvement of racemization, generality, and ligation reaction. In this view, we have developed a mild, practical, and efficient methods based on substrate-control by using boronic acid, niobium, and tantalum as catalysts. Our methods can be applied for a broad variety of amino acids to furnish the desired peptides in excellent yields without significant loss of stereochemical integrity. The developed straightforward approach overcome a lot of problem associated with peptide synthesis. This article describes our recent achievement based on catalytic substrate-controlled peptide synthesis.
Tomohiro Hattori*, Wataru Muramatsu*, Hisashi Yamamoto*
有機合成化学協会誌, 2021, 79, 382-390
Muramatsu, W. Manthena, C. Nakashima, E.
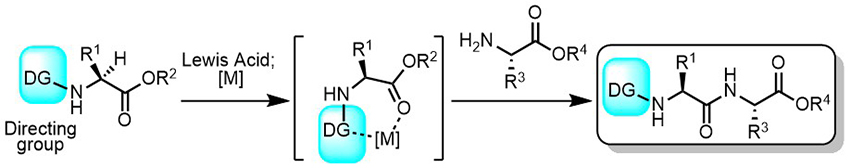
Peptide Bond-Forming Reaction via Amino Acid Silyl Esters: New Catalytic Reactivity of an Aminosilane
The epimerization-free formation of peptide bonds is crucial for the development of peptide therapeutics and pharmaceuticals. Herein, we report a hydrosilane-mediated approach for the construction of peptide bonds between most amino acids under ambient conditions. This concise protocol with an original silylating reagent HSi(OCH(CF3)2)3 facilitates the use of amino acids bearing a broad variety of functional groups without any epimerization. Moreover, a catalytic system using an aminosilane catalyst enables not only the acceleration in silylation of carboxylic acids but also amide synthesis with minimal substrate use (electrophile/nucleophile/silylating reagent = 1:1:1) and waste production (hydrogen gas and a siloxane). These simple and powerful approaches are established as a potentially general paradigm in synthesis of desired peptides in high yields.
Wataru Muramatsu*, Chaitanya Manthena, Erika Nakashima, Hisashi Yamamoto*
ACS Catal. 2020, 10, 9594-9603
Muramatsu, W. Hattori, T.

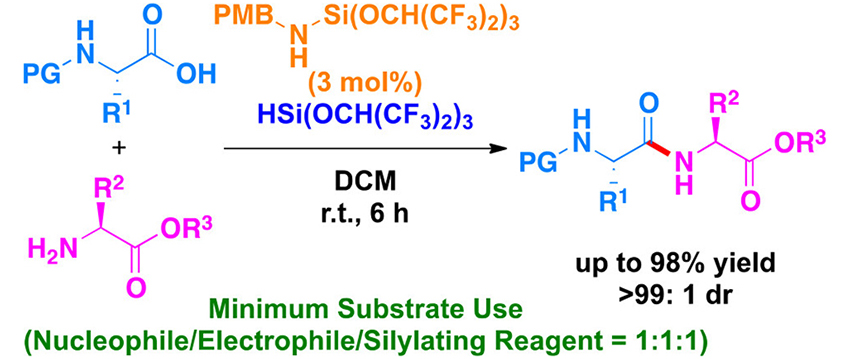
Game Change from Reagent- to Substrate-Controlled Peptide Synthesis
An account of the development of Lewis-acid-catalyzed methods for racemization-free peptide synthesis is presented. These methods are based on the substrate control concept that has been exploited extensively in stereoselective reactions, but the concept has never previously been applied to peptide synthesis. The most important difference that has emerged between our methods and the conventional methods based on reagent control concept such as coupling-reagent-mediated and boronic-acid-catalyzed peptide bond-forming reactions is how to activate the reaction sites and racemization control. The reagent-controlled methods proceed by generating highly reactive esters in situ, leading to occasional racemization through the formation of oxazolone intermediates. On the other hand, our substrate-controlled methods do not go through the known racemization processes because the Lewis acids we use herein are designed to activate moderately as an anchor a specific carbonyl group that is located at a reasonable distance from the directing group. Based on the substrate control concept, we have developed six novel methodologies for peptide bond-forming reactions over the last five years.
Wataru Muramatsu*, Tomohiro Hattori, Hisashi Yamamoto*
Bull. Chem. Soc. J. 2020, 6., 759-767
Sawano, T.

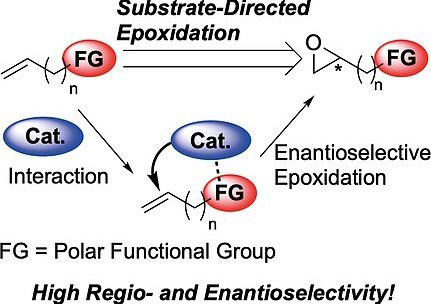
Regio- and Enantioselective Substrate-Directed Epoxidation
Substrate-directed reactionsare wellrecognized as important reactionstodistinguish sterically and electronically similar reactive sites based onpolarfunctional groupsto realize regioselective reactions.Sharplessasymmetric epoxidationis arepresentativesubstrate-directed reactionand known asanexceptionalreaction to provide usefulchiral epoxidesfrom allylic alcohols. Despite the considerable developmentofthe Sharpless asymmetric epoxidationover the lastfourdecades,somedifficult and challengingproblemsstill remainunsolved. Thisreviewdescribesthree fascinatingand attractive topics inenantioselective substrate-directedepoxidation:(1) carboxylic acid-or amine derivative-directedepoxidation,(2)regioselectiveepoxidationof non-proximalalkenes,(3)enantioselective epoxidationof bishomoallylic alcohols.
Takahiro Sawano*, Hisashi Yamamoto*
Eur. J. Org. Chem. 2020,2369-2378.
Muramatsu, W.

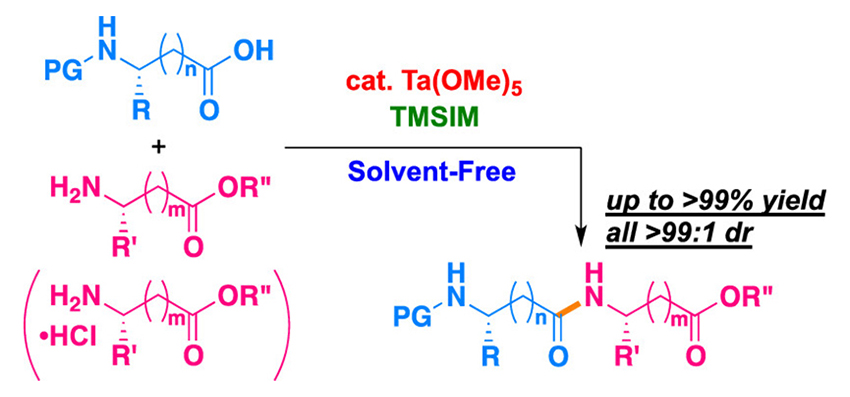
Tantalum-Catalyzed Amidation of Amino Acid Homologs
A tantalum-catalyzed solvent-free approach for the construction of amide bonds with 1-(trimethylsilyl)imidazole is developed, and the mild reaction conditions are applicable to a wide variety of electrophilic amino acid homologs. This approach delivers a new class of peptides in high yields without any epimerization.
Wataru Muramatsu*, Hisashi Yamamoto*
J. Am. Chem. Soc. 2019, 141, 18926-18931.
Muramatsu, W. Hattori T.

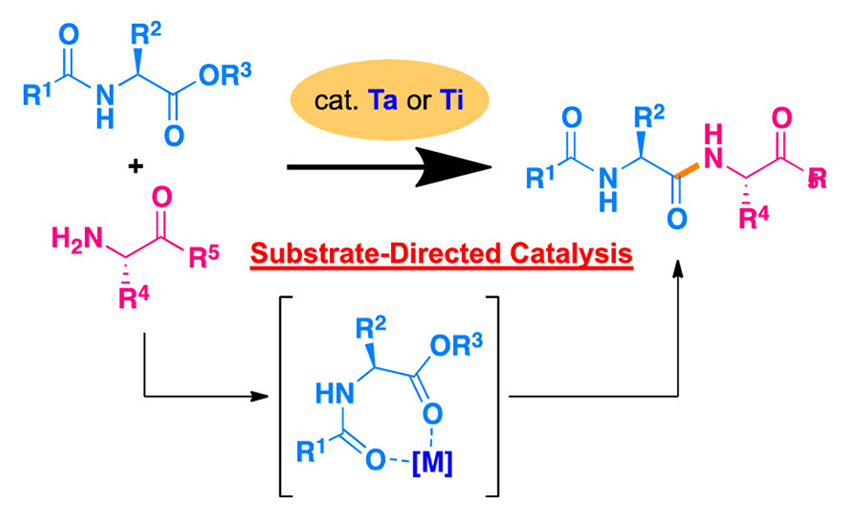
Substrate-Directed Lewis-Acids Catalysis for Peptide Synthsis
A Lewis-acid-catalyzed method for the substrate-directed formation of peptide bonds has been developed, and this powerful approach is utilized for the new “remote” activation of carboxyl groups under solvent-free conditions. The presented method has the following advantages: (1) the high-yielding peptide synthesis uses a tantalum catalyst for any amino acids; (2) the reaction proceeds without any racemization; (3) the new substrate-directed chemical ligation using the titanium catalyst is applicable to convergent peptide synthesis. These advantages overcome some of the unresolved problems in classical peptide synthesis.
Wataru Muramatsu*, Tomohiro Hattori and Hisashi Yamamoto*
J. Am. Chem. Soc. 2019, 141, 12288-12295.
Sawano, T

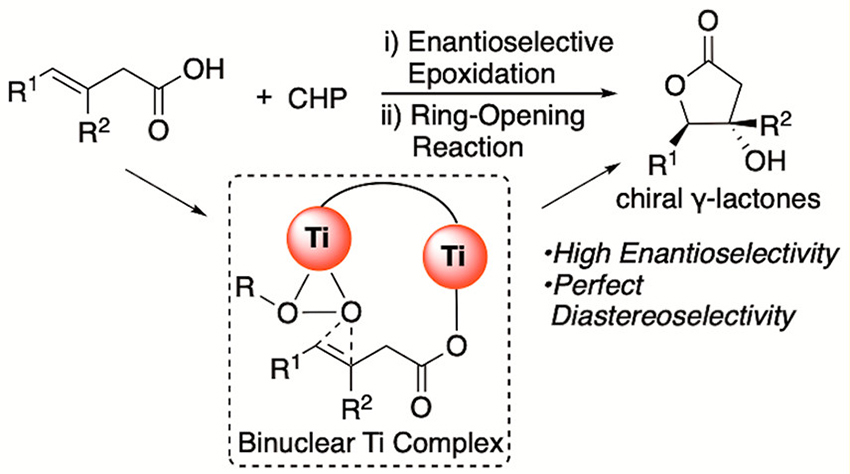
Enantioselective Epoxidation of β,γ-Unsaturated Carboxylic Acids by a Cooperative Binuclear Titanium Complex
Enantioselective epoxidation of β,γ-unsaturated carboxylic acids with a cooperative binuclear titanium complex has been developed to provide single diastereomer γ-lactones in high yields and enantioselectivities via intramolecular cyclization. For the success of the reaction, the proper distance between the carboxylic acid and alkene is quite important, offering the regioselective epoxidation of unsaturated carboxylic acids bearing two similar alkenes.
Takahiro Sawano* and Hisashi Yamamoto*
ACS catal. 2019, 9, 3384-3388.
Banerjee, A

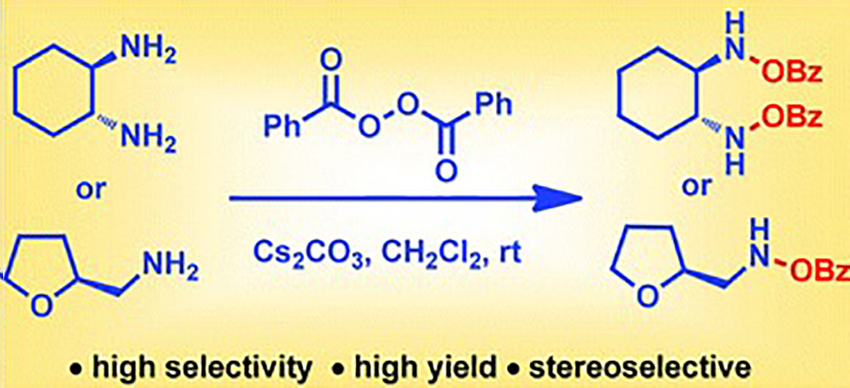
Direct N–O bond formation via oxidation of amines with benzoyl peroxide
Herein, we report a general and efficient method for direct N-O bond formation without undesirable C-N bond (amide) formation starting from commercially available amines and benzoyl peroxide. The oxidation of 1,2-diamines to furnish bis-(benzoyloxy)-1,2-diamines is reported for the first time. We found that a significant amount of water (BPO : water = 3 : 1) in combination with Cs2CO3 is necessary to achieve high selectivity and yield. The reaction conditions are applicable to a wide range of 1,2-diamine and 1,2-disubstituted-1,2-diamine substrates. Additionally this method is highly applicable to primary and secondary amines. Further, the present method can access chiral bis-hydroxamic acids and bis-hydroxyl amines in just two steps from 1,2-diamines. The reaction conditions are simple, mild and inert atmosphere free. The synthetic potential of this methodology is further demonstrated in the short synthesis of a chiral BHA ligand.
Amit Banerjee* and Hisashi Yamamoto*
Chem. Sci. 2019, 10, 2124-2129.
Bhadra, S.

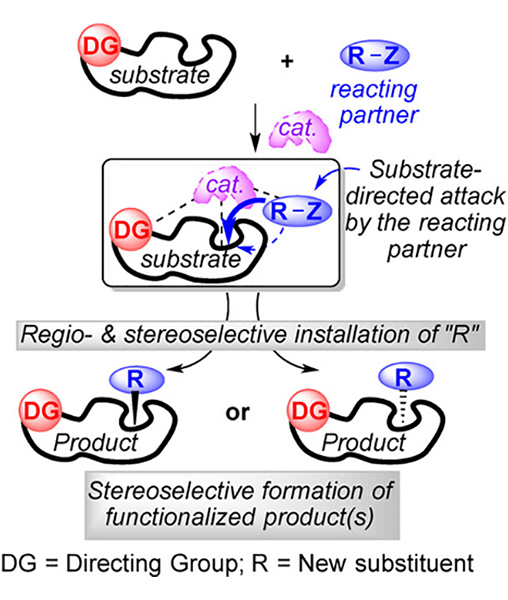
Substrate Directed Asymmetric Reactions
Historically, reagent controlled reactions (mechanism controlled reactions) have played a significant role in the asymmetric synthesis of complex structures. In contrast, today’s asymmetric synthesis is greatly dependent on substrate directed approaches. In this approach, a polar functional group, namely, a “directing group”, in the vicinity of the reactive site inside the substrate has been documented to preassociate with the chiral catalyst, which exerts stereodirecting influence by directing the reacting partner toward one of the enantiotopic faces of the reaction center. Those reactions usually proceed through exceptionally ordered transition states and result in extraordinary levels of stereoselection. Within the last four decades, the substrate directed approach has become an indispensible tool for the preparation of complex chiral frameworks starting directly from relatively simple achiral substrate molecules via asymmetric induction or various resolution techniques or both. Likewise, the substrate directed approach has been applied to functionalize enantiopure substrates bearing pre-exisiting stereocenters into complex structures as a single diastereomer. A classical example is Sharpless asymmetric epoxidation of allylic alcohols in which the free hydroxy function acts as an active anchor to a dimeric Ti-catalyst that controls the stereochemical outcome of the epoxidation process by transferring the oxidant enantioselectively. The principal aim of the present review is to give a general overview of substrate directed asymmetric transformations, a topic that has not yet been documented in the form of a concise review of recently developed approaches. Due to the large number of related applications, only recent advances that have been documented within the last two decades have been reviewed. Furthermore, in the current review, we have mainly highlighted asymmetric reactions that are controlled by abundant and frequently used directing groups such as hydroxy, amide, and sulfonamide groups. In addition, selected examples of a few important substrate-directed chemo-, regio-, and diastereoselective reactions have also been included in this review.
Sukalyan Bhadra* and Hisashi Yamamoto*
Chem. Rev. 2018, 118, 3391-3446.
Sawano, T.

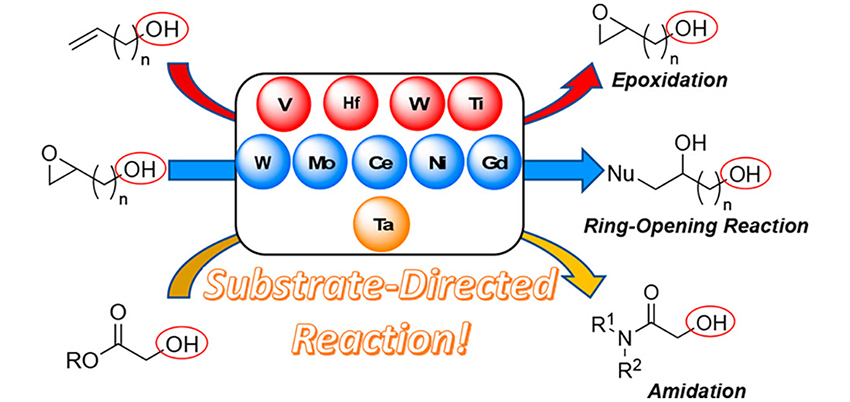
Substrate-Directed Catalytic Selective Chemical Reactions
The development of highly efficient reactions at only the desired position is one of the most important subjects in organic chemistry. Most of the reactions in current organic chemistry are reagent- or catalyst-controlled reactions, and the regio- and stereoselectivity of the reactions are determined by the inherent nature of the reagent or catalyst. In sharp contrast, substrate-directed reaction determines the selectivity of the reactions by the functional group on the substrate and can strictly distinguish sterically and electronically similar multiple reaction sites in the substrate. In this Perspective, three topics of substrate-directed reaction are mainly reviewed: (1) directing group-assisted epoxidation of alkenes, (2) ring-opening reactions of epoxides by various nucleophiles, and (3) catalytic peptide synthesis. Our newly developed synthetic methods with new ligands including hydroxamic acid derived ligands realized not only highly efficient reactions but also pinpointed reactions at the expected position, demonstrating the substrate-directed reaction as a powerful method to achieve the desired regio- and stereoselective functionalization of molecules from different viewpoints of reagent- or catalyst-controlled reactions..
Takahiro Sawano* and Hisashi Yamamoto*
J. Org. Chem. 2018, 83, 4889-4904.
Muramatsu, W.

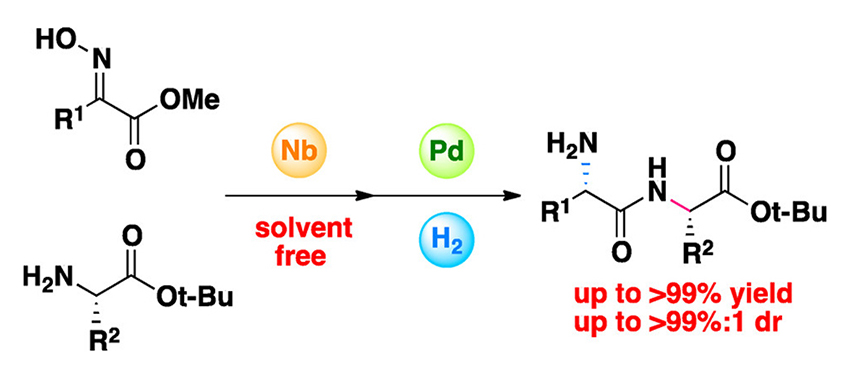
Catalytic Peptide Synthesis: Amidation of N‑Hydroxyimino Esters
A catalytic method for the formation of amide bonds was developed in which the amidation of N-hydroxyimino esters with a broad range of amino acid tert-butyl esters is promoted by a niobium catalyst in the absence of solvent. Contrary to the predominant protocol based on reagent control commonly applied to amidation reactions, this study provides insight into an approach based on substrate control. This system affords the corresponding amides in high yields in addition to being both highly atom efficient and racemization-free. An advantage of this system is shown in that the Lewis acid catalysis proceeds chemoselectively in the presence of nonactivated esters. Furthermore, the resulting amides are easily transformed into their corresponding di- and tripeptides with high diastereoselectivities under simple hydrogenation conditions.
Wataru Muramatsu* , Hiroaki Tsuji, and Hisashi Yamamoto*
ACS Catal. 2018, 8, 2181–2187
Nakashima, E.

Process Catalyst Mass Efficiency by Using Proline Tetrazole Column-Flow System
Generally, organocatalysts are not decomposed during chemical transformation, which is different from traditional metal catalysts. To improve catalytic processes efficiency, various studies have been applied to flow synthesis for organocatalysis. Furthermore, many immobilized organocatalysts have been used for heterogeneous flow synthesis, which requires huge amounts of immobilized catalyst and requires several steps to prepare. We took advantage of organocatalysts with low-polarity organic solvent and developed a flow system through a packed-bed column with simply proline tetrazole (5-(2-pyrrolidinyl)-1H-tetrazole) for heterogeneous organocatalytic synthesis. Under ambient temperature, this heterogeneous organocatalyst continuous flow-column system with ketones as a donor provides aldol, Mannich, and o-nitroso aldol reactions in up to quantitative yields with excellent enantio- and chemoselectivity values. Our heterogeneous-flow synthesis provides extremely low process catalyst mass efficiency and continuous production without changing the packed-bed catalyst column.
Erika Nakashima* and Hisashi Yamamoto*
Chem. Eur. J. 2018, 24, 1076 –1079.
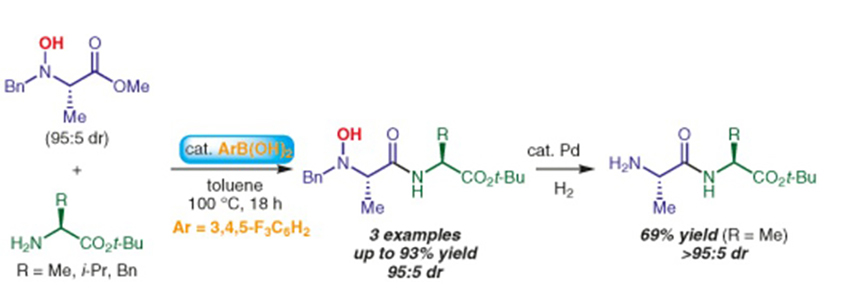
Tsuji, H.

Synthesis of Dipeptides by Boronic Acid Catalysis
We have found that a boronic acid catalyzed amidation of an N-hydroxy amino acid methyl ester with amino acid tert-butyl esters gave N-hydroxy dipeptide derivatives in good yields without any racemization. The protecting groups on the nitrogen atom could be easily removed by heterogeneous hydrogenation conditions.
Hiroaki Tsuj* and Hisashi Yamamoto*
Synlett. 2018, 29, 318–321
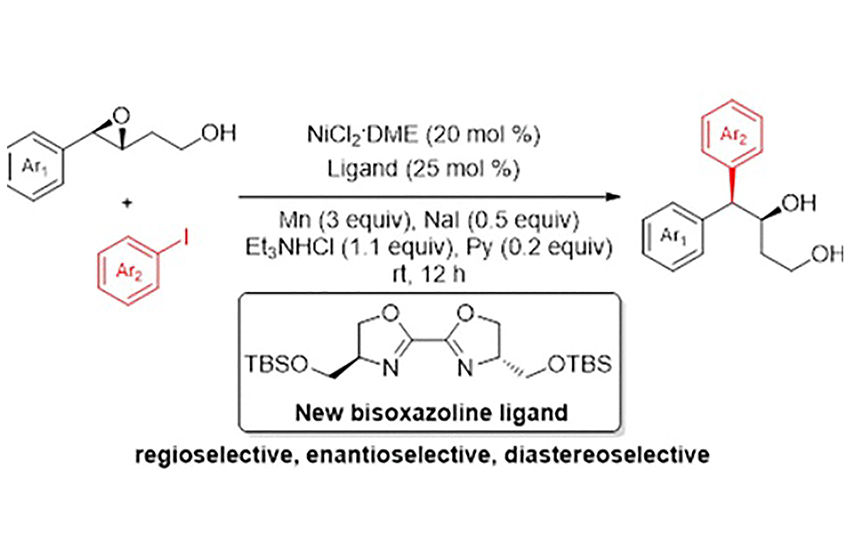
Banerjee, A

Nickel Catalyzed Regio-, Diastereo-, and Enantioselective Cross-Coupling of 3,4-Epoxyalcohols with Aryl Iodides
The first catalytic, regioselective cross-coupling of 3,4-epoxyalcohol with aryl iodides is reported. The combination of NiCl2·DME and a newly developed C2-symmetric oxazoline ligand plays a key role in selective ring opening of several 3,4-epoxy alcohols at the C4 position. This general protocol furnishes a new type of enantioenriched 4,4-diaryl alkane which also incorporates an additional 1,3-diol that can be easily transformed to a variety of functional groups. The products are formed with excellent regioselectivity (>99:1), diastereoselectivity (up to 99:1), and enantiopurity (up to >99.9% ee).
Amit Banerjee*, Hisashi Yamamoto*
Org Lett. 2017,19, 4363-4366.
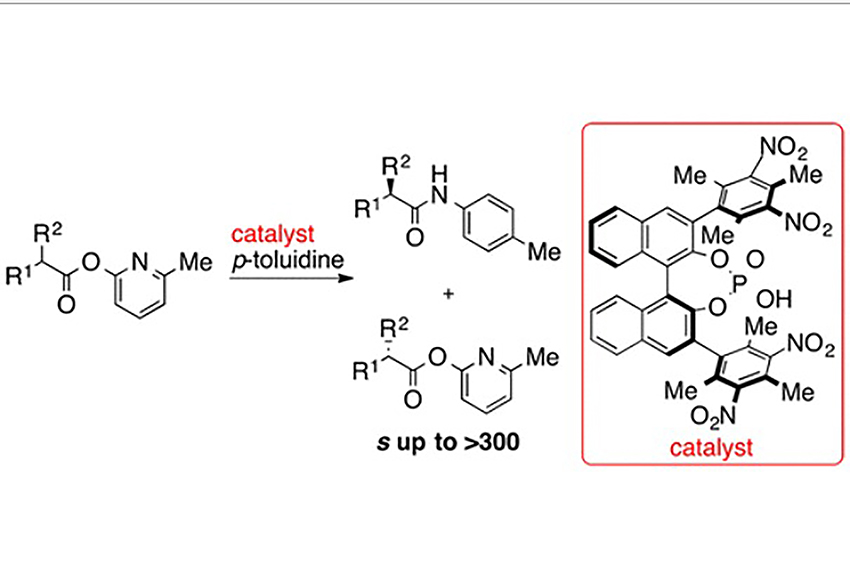
Shimoda, Y

Chiral Phosphoric Acid-Catalyzed Kinetic Resolution via Amide Bond Formation
We describe the kinetic resolution of a readily available 2-pyridyl ester via an amide bond formation catalyzed by a chiral Brønsted acid. A chiral phosphoric acid bearing a 2,4,6-trimethyl-3,5-dinitrophenyl group at the 3,3′-position enabled this transformation with high selectivities. We also found that the addition of Lewis acid increased both the reactivity and selectivity in the substrate with a methoxy group.
Yasushi Shimoda*, Hisashi Yamamoto*
J. Am. Chem. Soc. 2017, 139, 6855-6858
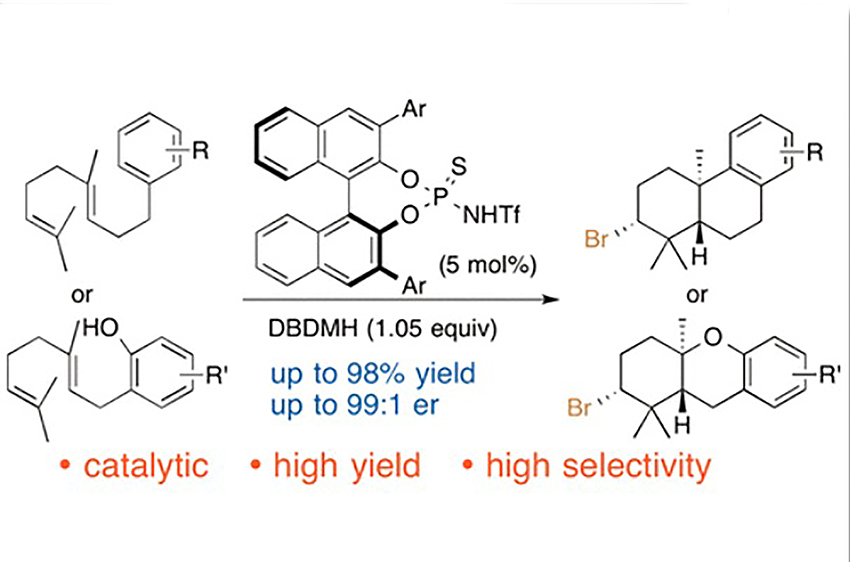
Samanta, R. C.

Catalytic Asymmetric Bromocyclization of Polyenes
The first catalytic asymmetric bromonium ion-induced polyene cyclization has been achieved by using a chiral BINOL-derived thiophosphoramide catalyst and 1,3-dibromo-5,5-dimethylhydantoin as an electrophilic bromine source. Bromocyclization products are obtained in high yields, with good enantiomeric ratios and high diastereoselectivity, and are abundantly found as scaffolds in natural products.
Ramesh C. Samanta*, Hisashi Yamamoto*
J. Am. Chem. Soc. 2017, 139, 1460-1463
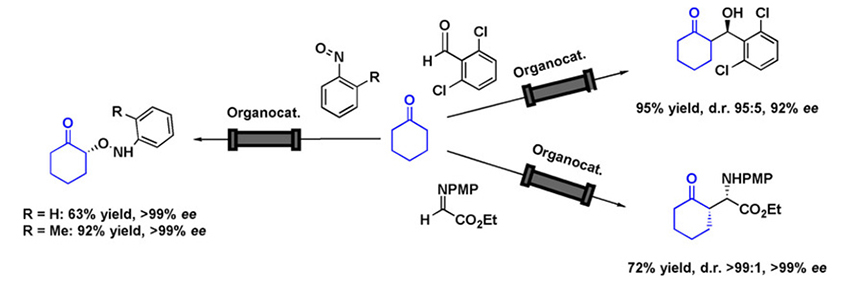
Nakashima, E.

Asymmetric Aldol Synthesis: Choise of Organocatalyst and Conditions
Proline tetrazole showed an excellent catalytic activity for the aldol reaction between acetone and 4-nitrobenzaldehyde at 0.1 mol % catalyst loading. Using water as an additive led to excellent yields and high enantioselectivities, even on a 0.1 mol-scale production (>99 % isolated yield, 84 % ee).
Erika Nakashima*, Hisashi Yamamoto*
Chem. Asian. J. 2017, 12, 41-44.

Tsuji, H

Hydroxy-Directed Amidation of Carboxylic Acid Esters Using a Tantalum Alkoxide Catalyst
We describe herein a new strategy for the chemoselective synthesis of amides by using a metal-catalyzed hydroxy-directed reaction. A hydroxy group located at the β-position of an ester group promoted the activation of a carbonyl group with a tantalum alkoxide catalyst followed by amidation reactions, leading to a wide variety of β-hydroxyamides with excellent chemeselectivity. The chemoselective amidation strategy can be extended to the catalytic synthesis of dipeptide derivatives, which remains challenging research subjects in modern organic synthesis.
Hiroaki Tsuji*, Hisashi Yamamoto*
J. Am. Chem. Soc. 2016, 138, 14218-14221
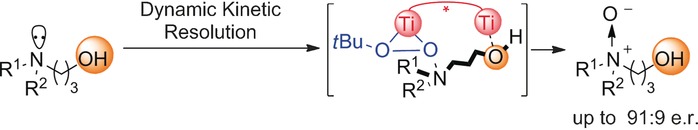
Bhadra, S.

Catalytic Asymmetric Synthesis of N-Chiral Amine Oxides
An asymmetric synthesis of N-chiral amine oxides was developed that proceeds via dynamic kinetic resolution of unsymmetrical γ-hydroxy tertiary amines. A binuclear titanium complex enables the desired hydroxy-directed N-oxidation process. The concept was further expanded to the kinetic resolution of racemic γ-amino alcohols with a preexisting stereocenter.
Sukalyan Bhadra*, Hisashi Yamamoto*
Angew. Chem. Int. Ed. 2016, 55, 13043-13046
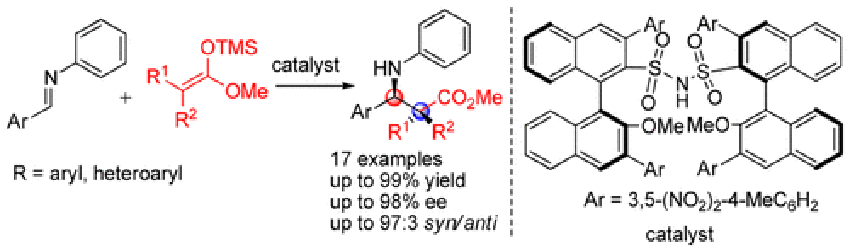
Zhou, F.
A Disulfonimide Catalyst for Highly Enantioselective Mukaiyama-Mannich Reaction
A new BINOL-derived chiral disulfonimide has been developed by introducing 4-methyl-3,5-dinitrophenyl substituents at its 3- and 3′-positions. This chiral disulfonimide catalyst displays high catalytic efficacy toward the asymmetric Mukaiyama–Mannich reaction of imines with ketene silyl acetals leading to β-amino acid esters in good yields (up to 99%) with high diastereoselectivities (syn/anti up to 97:3) and enantioselectivities (up to 98% ee). The long-standing problem of the chiral phosphoric acid-catalyzed asymmetric Mukaiyama–Mannich reaction that requires a 2-hydroxyphenyl moiety was solved by this disulfonimide catalyst.
Fengtao Zhou*, Hisashi Yamamoto*
Org. Lett. 2016, 18, 4974-4977.
Gati, W.
Second Genaration of Aldol Reaction
Since the discovery of the Mukaiyama aldol reaction more than 40 years ago, several landmark publications have inspired researchers in the field. The Mukaiyama AR is one of the most significant named reactions in organic synthesis. In the past few decades, development of the modern AR has been at the forefront in addressing the challenges of regio-, chemo-, diastereo-, and enantioselectivity in organic synthesis. All of these selectivity challenges maybe present in a single pair of reactants, thus controlling the outcome of such a process has great practical value. More than 10 years ago, our group became involved in this iconic carbon–carbon bond-forming process and attempted to very closely investigate all possible features of the AR to solve several issues still encountered by chemists, most notably the selectivity challenges mentioned above. In this context, our group initiated the second generation of the AR based on a Lewis or Brønsted acid-catalyzed process in conjunction with the use of a “super silyl” (tris(trimethylsilyl)silyl) directing group, which has demonstrated unrivalled properties in controlling the outcome of the AR. Using the extraordinary power of the super silyl group, we were able to develop new methods and concepts that broadly impacted the ability to control the selectivity attributes and thus allowed for a highly stereoselective construction of polyketide, halogenated polyketide, polypropionate, and polyol scaffolds through inter- and/or intramolecular aldolization protocols. Our diastereoselective ARs of super silyl enol ethers and aldehydes have shown great efficiency and modularity in producing exclusively and preferentially syn- or anti-adducts, creating up to four new adjacent stereocenters in a one-pot sequential manner and under mild reaction conditions. The super silyl-directed AR does not only provide a solution to stereochemistry control challenges, but also offers an efficient, modular and high yielding technique toward nontrivial construction of complex architectures with unprecedented ease. We believe that the new Lewis- or Brønsted-acid-catalyzed super-silyl-directed AR processes chronicled in our laboratories have come to maturity and now offer a “road map” for strategic stereoselective synthesis of polyketide-like units. Herein we report our recent achievements in the diastereoselective C–C bond formation, through the super-silyl-directed AR, toward the synthesis of complex and sophisticated hydroxy aldehydes. We would like to note that due to the extremely broad range of work reported in this field, only stereoselective AR involving aldehyde-derived super SEEs will be discussed in this Account.
Wafa Gati*, Hisashi Yamamoto*
Acc. Chem. Res. 2016, 49, 1757-1768.
Zhou, F.
A Powerful Chiral Phosphoric Acid Catalyst for Enantioselective Mukaiyama–Mannich Reactions
A new BINOL-derived chiral phosphoric acid bearing 2,4,6-trimethyl-3,5-dinitrophenyl substituents at the 3,3′-positions was developed. The utility of this chiral phosphoric acid is demonstrated by a highly enantioselective (ee up to >99 %) and diastereoselective (syn/anti up to >99:1) asymmetric Mukaiyama–Mannich reaction of imines with a wide range of ketene silyl acetals. Moreover, this method was successfully applied to the construction of vicinal tertiary and quaternary stereogenic centers with excellent diastereo- and enantioselectivity. Significantly, BINOL-derived N-triflyl phosphoramide constitutes a complementary catalyst system that allows the title reaction to be applied to more challenging imines without an N-(2-hydroxyphenyl) moiety.
Fengtao Zhou*, Hisashi Yamamoto*
Angew. Chem. Int. Ed. 2016, 55, 8970-8974.
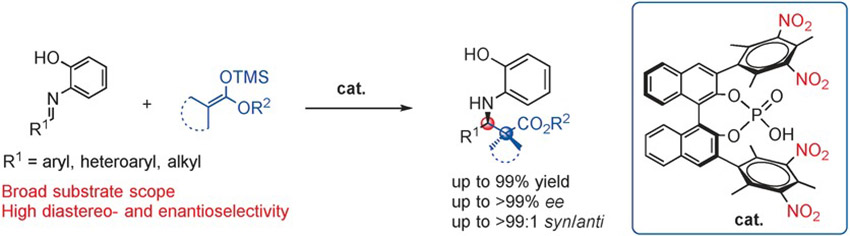
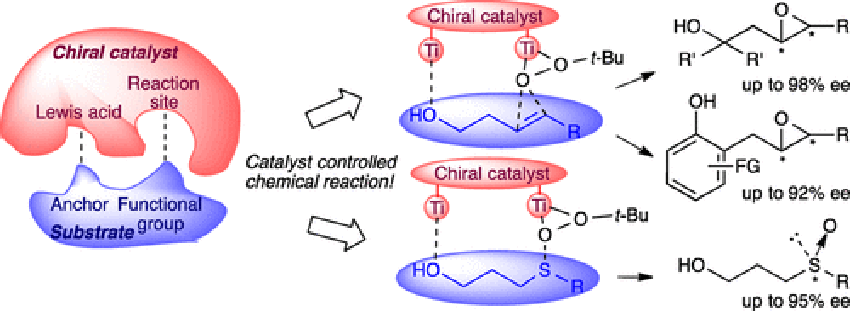
Bhadra, S.

Design of a New Bimetallic Catalyst for Asymmetric Epoxidation and Sulfoxidation
A new chiral tethered 8-quinolinol-based ligand class is developed. The binuclear titanium complex of the ligand operates through a novel mechanism allowing for the regio- and stereoselective epoxidation of primary and tertiary homoallylic alcohols (up to 98% ee), as well as first examples of 2-allylic phenols (up to 92% ee). The new catalyst system also promotes the asymmetric oxidation of γ-hydroxypropyl sulfides giving an important class of chiral sulfoxides that have been inaccessible to date (up to 95% ee).
Sukalyan Bhadra*, Matsujiro Akakura, Hisashi Yamamoto*
J. Am. Chem. Soc. 2016, 137, 15612-15615.
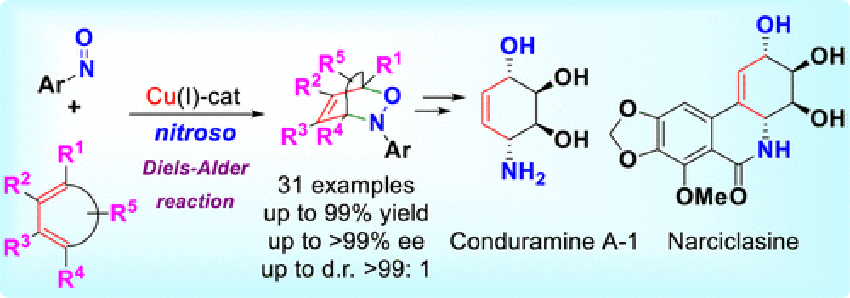
Maji, B.
Catalytic Enantioselective Nitroso Diels-Alder Reaction
The nitroso Diels–Alder (NDA) reaction is an attractive strategy for the synthesis of 3,6-dihydro-1,2-oxazines and 1-amino-4-hydroxy-2-ene derivatives. Herein we report the Cu(I)–DTBM-Segphos catalyzed asymmetric intermolecular NDA reaction of variously substituted cyclic 1,3-dienes using highly reactive nitroso compounds derived from pyrimidine and pyridazine derivatives. In most of the cases studied, the cycloadducts were obtained in high yields (up to 99%) with very high regio-, diastereo-, and enantioselectivities (up to regioselectivity > 99:1, d.r. > 99:1, and >99% ee). As an application of this methodology, formal syntheses of conduramine A-1 and narciclasine were accomplished.
Biplab Maji*, Hisashi Yamamoto*
J. Am. Chem. Soc. 2015, 137, 15957-15963.
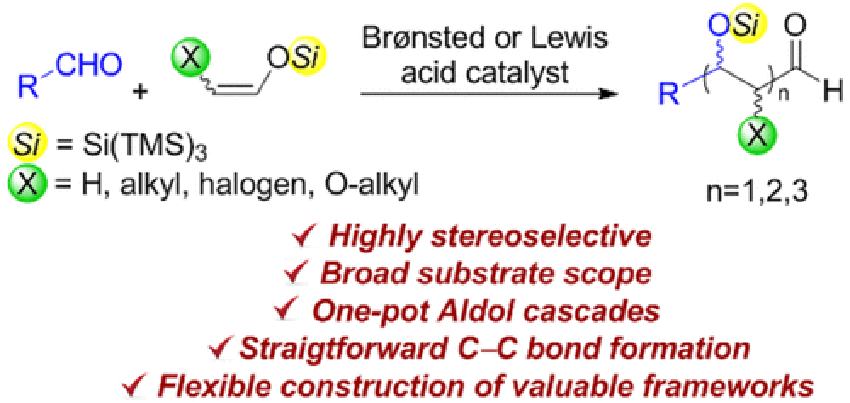
Gati, W.
A highly diastereoselective “super silyl” governed aldol reaction: synthesis of α,β-dioxyaldehydes and 1,2,3-triols
A highly diastereoselective approach for the synthesis of protected α,β-dioxyaldehydes derived from (Z)-tris(trimethylsilyl)silyl “super silyl” enol ethers is described. A general and highly syn-stereoselective aldol reaction directed by the “super silyl” group catalyzed by triflimide (HNTf2) is developed providing α,β-dioxyaldehydes and 1,2,3-triol fragments which can be a useful platform for the elaboration of natural and unnatural sugar derivatives.
Wafa Gati*, Hisashi Yamamoto*
Chem. Sci 2016, 7, 394-399.
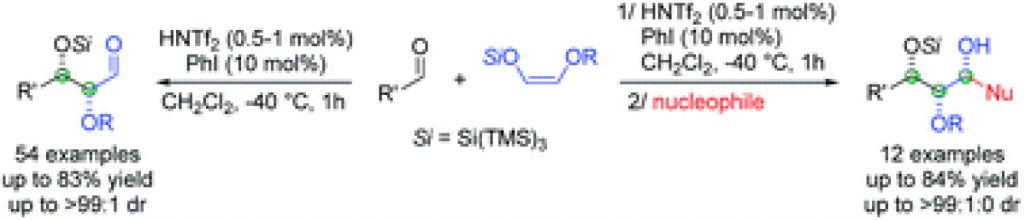
Nakashima, E.

Continuous flow of Nitroso Diels-alder reaction
Our flow reaction systems have provided quantitative yields of nitroso Diels–Alder products with no byproducts in cases of cyclic dienes without temperature and pressure controls. Additionally, the reaction times were significantly shortened by using homogeneous catalyst (CuCl) or heterogeneous reagent (MnO2) in comparison with batch reaction.
Erika Nakashima*, Hisashi Yamamoto*
Chem. Commun. 2015, 51, 12309-12312
Samanta, R. C.

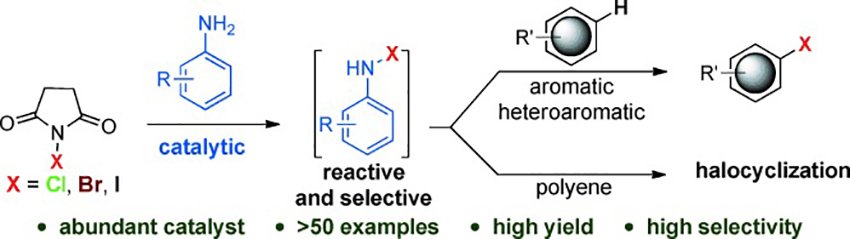
Selective Halogenation Reaction Using An Aniline Catalyst
Electrophilic halogenation is used to produce a wide variety of halogenated compounds. Previously reported methods have been developed mainly using a reagent-based approach. Unfortunately, a suitable “catalytic” process for halogen transfer reactions has yet to be achieved. In this study, arylamines have been found to generate an N-halo arylamine intermediate, which acts as a highly reactive but selective catalytic electrophilic halogen source. A wide variety of heteroaromatic and aromatic compounds are halogenated using commercially available N-halosuccinimides, for example, NCS, NBS, and NIS, with good to excellent yields and with very high selectivity. In the case of unactivated double bonds, allylic chlorides are obtained under chlorination conditions, whereas bromocyclization occurs for polyolefin. The reactivity of the catalyst can be tuned by varying the electronic properties of the arene moiety of catalyst.
Ramesh C. Samanta*, Hisashi Yamamoto*
Chem. Eur. J. 2015, 21, 11976-11979
Izumiseki, A.
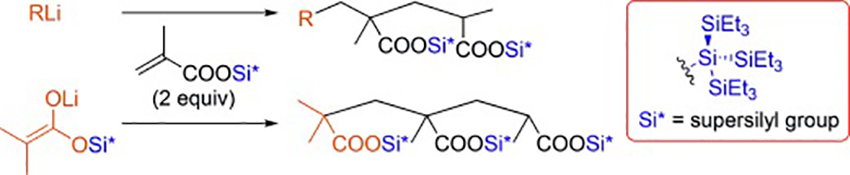
Selective Michael Reaction Controlled by Supersilyl Protecting Group
Selective Michael reaction of organolithium reagents to supersilyl methacrylate is reported. The method was able to control a single and double Michael addition. The successful termination of the process using the supersilyl protecting group allows for the controlled, chemoselective, and diastereoselective Michael reaction.
Atsuto Izumiseki*, Hisashi Yamamoto*
Angew. Chem. Int. Ed. 2015. 54, 8697-8699.
Sai, M.
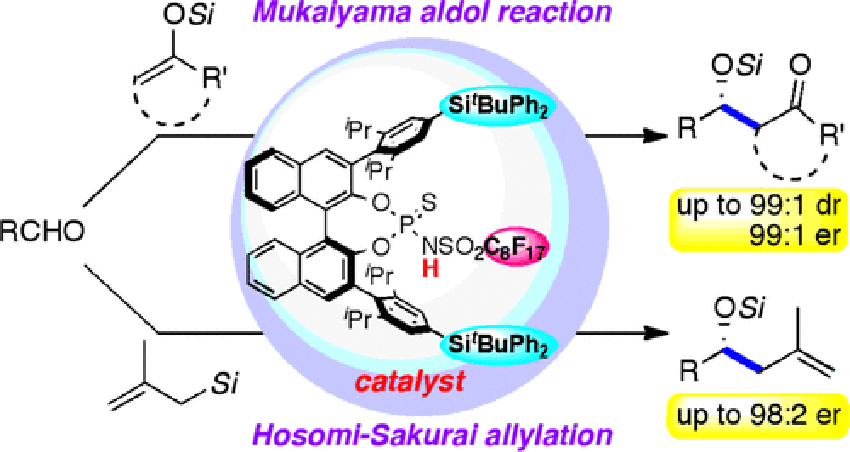
Chiral Brønsted Acid as a True Catalyst: Asymmetric Mukaiyama Aldol and Hosomi-Sakurai Allylation Reactions
Highly diastereo- and enantioselective Mukaiyama aldol reaction catalyzed by a new chiral Brønsted acid, N-(perfluorooctanesulfonyl)thiophosphoramide, is described. The perfluorooctyl substituent on the sulfonyl group of the catalyst plays an essential role in the stereoselection. The catalyst also allows the asymmetric Hosomi–Sakurai allylation, which has been considerably challenging due to the low reactivity of allylsilanes. 29Si and 31P NMR monitoring reveals the characteristic feature of the thiophosphoramide catalyst, acting as a strong Brønsted acid even in the presence of excess silyl nucleophiles, which cannot be found in other related phosphoric acid analogues.
Masahiro Sai*, Hisashi Yamamoto*
J. Am. Chem. Soc.2015. 137, 7091-7094.
Maji, B.
Use of In Situ Generated Nitrosocarbonyl Compounds in Catalytic Asymmetric α-Hydroxylation and α-Amination Reactions
The in situ generation of nitrosocarbonyl compound from N-protected hydroxylamines via oxidation using MnO2 as oxidant is compatible with Lewis- and organocatalyzed processes. Thus highly functionalized tertiary α-hydroxy and α-amino carboxylic acids, α-hydroxy phosphonic acids and β-amino alcohols were synthesized.
Biplab Maji*, Hisashi Yamamoto*
BCSJ. 2015. in press.
Maji, B.
Copper-Catalyzed Asymmetric Synthesis of Tertiary α-Hydroxy Phosphonic Acid Derivatives with In Situ Generated Nitrosocarbonyl Compounds as the Oxygen Source
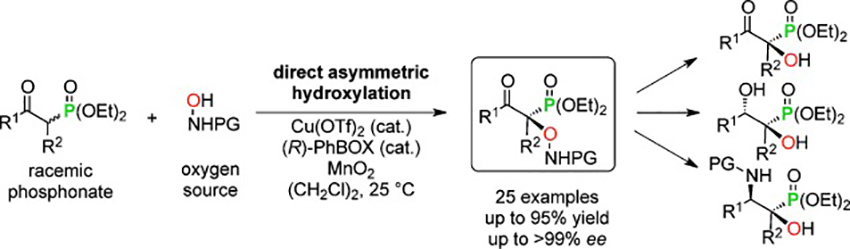
α-Hydroxy phosphonic acids and their derivatives are highly bioactive structural motifs. It is now reported that these compounds can be accessed through the copper-catalyzed direct α-oxidation of β-ketophosphonates using in situ generated nitrosocarbonyl compounds as an electrophilic oxygen source. These reactions proceeded in high yields (up to 95 %) and enantioselectivities (up to >99 % ee) for both cyclic as well as acyclic substrates. This method was also applied for the synthesis of α,β-dihydroxy phosphonates and β-amino-α-hydroxy phosphonates.
Biplab Maji*, Hisashi Yamamoto*
Angew. Chem. Int. Ed. 2014. 53, 14472-14475.
Selected as a Hot paper.
Sai, M.
Chemoselective silyl transfer in the Mukaiyama aldol reaction promoted by super silyl Lewis acid
In the silyl Lewis acid-promoted Mukaiyama aldol reaction, the steric and electronic properties of the silyl group on the silyl Lewis acid influence the reaction mechanism and product distribution. When super silyl triflates such as (TMS)3SiOTf and (TES)3SiOTf are used as Lewis acids, the silyl group of the silyl enol ether chemoselectively transfers to the product. The mechanistic details have been investigated using density functional theory (DFT) calculations.
Masahiro Sai*, Matsujiro Akakura, Hisashi Yamamoto*
Chem. Commun. 2014. 50, 15206-15208.
Maji, B.
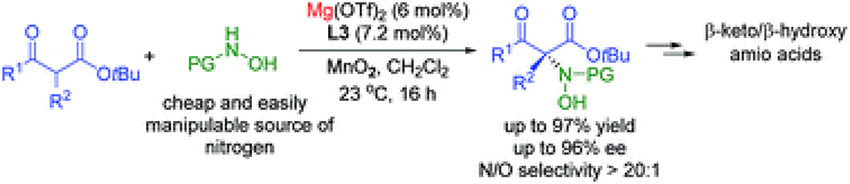
Asymmetric construction of quaternary stereocenters by magnesium catalysed direct amination of β-ketoesters using in situ generated nitrosocarbonyl compounds as nitrogen sources
The first example of a Lewis acid catalysed asymmetric hydroxyamination of β-ketoesters with nitrosocarbonyl compounds generated in situ was accomplished. The combination of a catalytic amount of Mg(OTf)2 with a chiral N,N‘-dioxide ligand provides highly substituted quaternary β-keto amino acid derivatives in high yields (up to 97%) and enantioselectivities (up to 96%). Regioselectivities (N- vs. O-attack) are uniformly high for all substrates (>20 : 1).
Biplab Maji*, Mahiuddin Baidya, Hisashi Yamamoto*
Chem. Sci. 2014. 5. 3941-3945.
Maji, B.
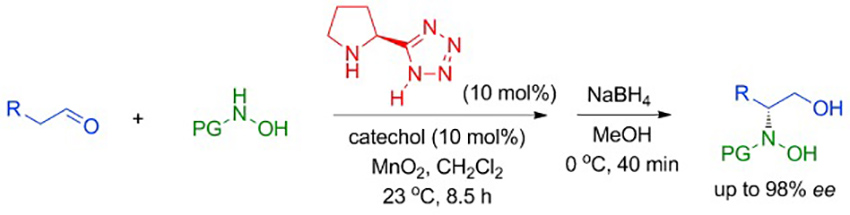
Proline-Tetrazole-Catalyzed Enantioselective N-Nitroso Aldol Reaction of Aldehydes with In Situ Generated Nitrosocarbonyl Compounds
A highly enantioselective method (up to 98 % ee) for the preparation of β-amino alcohols was achieved by using the readily available proline-tetrazole as the catalyst for the N-nitroso aldol reaction of aldehydes with in situ generated nitrosocarbonyl compounds. The key to success of this reaction is the use of MnO2 as an oxidant and catechol as a Brønsted acid additive.
Biplab Maji*, Hisashi Yamamoto*
Angew. Chem. Int. Ed. 2014. 53. 8714-8717.
Highlighted in Synfacts, 2014. 10. 0531.
Izumiseki, A.
Intermolecular/Intramolecular Sequential Aldol Reaction
The first example of intermolecular/intramolecular sequential aldol reaction of disilyl enol ethers is described. This strategy enables the formation of five-, six- and seven-membered ring products. Four or more contiguous stereogenic centers are created with high levels of relative stereochemical control.
Atsuto Izumiseki*, Hisashi Yamamoto*
J. Am. Chem. Soc. 2014. 136. 1308-1311.
Highlighted in Synfacts, 2014. 10. 0503.